
查疾病
您的当前位置:首页 > 查疾病 > 重症肌无力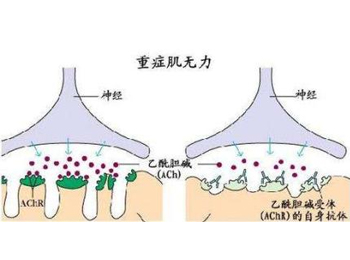
重症肌无力(myasthenia gravis,MG)是一种横纹肌神经肌肉接头点处传导障碍的自身免疫性疾病,以肌肉易疲劳晨轻暮重、休息或用胆碱酯酶抑制药后减轻为特点。常累及眼外肌、咀嚼肌、吞咽肌和呼吸肌。严重者球麻痹。受累肌肉的分布因人因时而异,而并…
查看详细早期症状:颈肌、躯干及四肢肌也可罹病,表现抬头困难,常用手托住头颅;胸闷气短,行走乏力,不能久行;洗脸、梳头、穿衣难于支持;腱反射存在,无感觉障碍;呼吸肌、膈肌受累, 出现咳嗽无力、呼吸困难
晚期症状:可因呼吸肌麻痹&继发吸入性肺炎可导致死亡。偶心肌受累可突然死亡,平滑肌和膀胱括约肌一般不受累;严重时出现肢体无力, 上肢重于下肢, 近端重于远端。偶见肌萎缩。
相关症状:无力、面肌无力、疲劳、吞咽困难、呼吸困难
一、症状:
本病可见于任何年龄,我国病人发病年龄以儿童期较多见,20~40岁发病者女性较多,中年以后发病者多为男性,伴有胸腺瘤的较多见。女性病人所生新生儿,其中约10%经过胎盘转输获得烟碱型乙酰胆碱受体抗体(nicotiniacetylcholine receptorantibody,nAchR-Ab)可暂时出现无力症状。少数有家族史。起病隐袭,也有急起暴发者。
1.传统的分型:眼肌型、延髓肌型和全身型。
(1)眼肌型重症肌无力:临床特征是受累骨骼肌肉呈病态疲劳,症状多在下午或傍晚劳累后加重,早晨和休息后减轻,呈规律的晨轻暮重波动性变化。肌无力通常晨轻晚重,亦可多变,后期可处于不全瘫痪状态;全身肌肉并非平均受累,眼外肌最常累及,为早期症状,亦可长期局限于眼肌。轻者睁眼无力,眼睑下垂,呈不对称性分布,额肌代偿性地收缩上提。眼球运动受限,出现斜视和复视,重者眼球固定不动。眼内肌一般不受影响,瞳孔反射多正常。
(2)延髓型(或球型)重症肌无力:面肌、舌肌、咀嚼肌及咽喉肌亦易受累;闭眼不全,额纹及鼻唇沟变平,笑时口角后缩肌比上唇提肌更无力,提唇露齿如怒吼状;咀嚼无力,吞咽困难,舌运动不自如;软腭肌无力,发音呈鼻音;谈话片刻后音调低沉或声嘶。
(3)全身型重症肌无力:颈肌、躯干及四肢肌也可罹病,表现抬头困难,常用手托住头颅;胸闷气短,行走乏力,不能久行;洗脸、梳头、穿衣难于支持;腱反射存在,无感觉障碍;呼吸肌、膈肌受累, 出现咳嗽无力、呼吸困难,重症可因呼吸肌麻痹&继发吸入性肺炎可导致死亡。偶心肌受累可突然死亡,平滑肌和膀胱括约肌一般不受累;严重时出现肢体无力,上肢重于下肢,近端重于远端。偶见肌萎缩。
以上分型并非绝然分隔,往往混杂存在,而以某类症状较为突出。本病病程稽延,其间可缓解、复发或恶化。感冒、腹泻、激动、过劳及月经、分娩或手术等常使病情加重,甚至出现危象危及生命。奎宁, 奎尼丁, 普鲁卡因酰胺, 青霉胺, 心得安, 苯妥英, 锂盐, 四环素及氨基糖甙类抗生素可加重症状,避免使用。临床检查受累肌易疲劳,持续活动导致暂时性肌无力加重, 短期休息后好转是MG特征性表现;受累肌无力不符合任单一神经、神经根或中枢神经系统病变分布;进展性病例受累肌可轻度肌萎缩, 感觉正常, 通常无反射改变 。
临床分型:
1.Osserman分型:
Ⅰ型:眼肌型(15~20%),单纯眼外肌受累。症状主要是单纯眼外肌受累,表现为一侧或双侧上睑下垂,有复视或斜视现象。肾上腺皮质激素治疗有效,预后好。
Ⅱ型:全身型,累及一组以上延髓支配的肌群,病情较I型重,累及颈、项、背部及四肢躯干肌肉群。据其严重程度可分为Ⅱa与Ⅱb型。
ⅡA型:轻度全身型(30%),进展缓慢,无危象,常伴眼外肌受累,无咀嚼、吞咽及构音障碍,下肢无力明显,登楼抬腿无力,无胸闷或呼吸困难等症状。对药物反应好,预后较好。
ⅡB型:中度全身型(25%),骨骼肌和延髓肌严重受累,明显全身无力,生活尚可自理,伴有轻度吞咽困难,时有进流汁不当而呛咳,感觉胸闷,呼吸不畅。
无危象,药物敏感欠佳。
Ⅲ型:重症急进型(15%),症状危重,进展迅速,数周或数月内达到高峰,胸腺瘤高发。可发生危象,药效差,常需气管切开或辅助呼吸,死亡率高。
Ⅳ型:迟发重症型(10%),症状同Ⅲ型,从Ⅰ型发展为ⅡA、ⅡB型,经2年以上进展期,逐渐发展而来。药物治疗差,预后差。
Ⅴ型:肌萎缩型,起病半年出现肌肉萎缩,生活不能自理,吞咽困难,食物误入气管而由鼻孔呛出。口齿不清或伴有胸闷气急。因长期肌无力而出现继发性肌萎缩者不属于此型。病程反复2年以上,常由Ⅰ型或Ⅱ型发展而来。
二、疾病早期体征:
包括眼睑下垂、复视、说话费力、吞咽困难和轻度肢体肌无力等,脑神经支配肌持续活动后出现疲劳,如凝视天花板可加重眼睑下垂,凝视或阅读2~3min后出现复视,稍事休息后可恢复。
为部分或全身骨骼肌易疲劳,波动性肌无力,活动后加重、休息后减轻和晨轻暮重特点,体检无其他神经系统体征,低频重复电刺激波幅递减,胆碱酯酶抑制药治疗有效和对箭毒类药物超敏感等药理学特点,以及血清AchR-Ab增高等。
三、其他分类:
(1)新生儿MG:约12%MG母亲的新生儿有吸吮困难、哭声无力、肢体瘫痪,待别是呼吸功能不全的典型症状。症状出现在生后48h内,可持续数日至数周,症状进行性改善,直至完全消失。母亲、患儿都能发现AchR—Ab,症状随抗体滴度降低而消失。血浆置换可用于治疗严重呼吸功能不全患儿,呼吸机支持和营养也是治疗的关键。
(2)先天性MG:少见,但症状严重。通常在新生儿期无症状,婴儿期主要症状是眼肌麻痹、肢体无力亦明显,有家族性病史。AchR—Ab阴性,但重复电刺激反应阳性。已知AchR基因突变所产生的离子通道疾病有慢通道综合征和快通道综合征。前者为离子通道开放期异常延长而使其对Ach反应增强,尤使前臂伸肌肌力选择性减弱,奎尼丁有疗效,可使延长的开放期缩短;后者对Ach反应减弱。己知AChR w亚单位的24种突变都是常染色体隐性遗传、造成终板AchR的严重缺失。抗胆碱酯酶药物可能有效。
(3)药源性MG:可发生在用青霉胺治疗类风湿关节炎、硬皮病、肝豆状核变性的患者。临床症状和AchR—Ab滴度与成人型MG相似,停药后症状消失。
四、其他:危象
是指肌无力突然加重,特别是呼吸肌(包括膈肌、肋间肌)以及咽喉肌的严重无力,导致呼吸困难,喉头与气管分泌物增多而无法排出,需排痰或人工呼吸。多在重型肌无力基础上诱发,伴有胸腺瘤者更易发生危象。危象可分为3种:
(1)肌无力危象 为疾病本身肌无力的加重所致,此时胆碱酯酶抑制药往往药量不足,加大药量或静脉注射腾喜龙后肌力好转。常由感冒诱发,也可发生于应用神经-肌肉阻滞药(如链霉素)、大剂量皮质类固醇及胸腺放射治疗或手术后。
(2)胆碱能危象 是由于胆碱酯酶抑制药过量,使Ach免于水解,在突触积聚过多,表现胆碱能毒性反应;肌无力加重、肌束颤动(烟碱样反应,终板膜过度除极化);瞳孔缩小(于自然光线下直径小于2mm)、出汗、唾液增多(毒蕈碱样反应),头痛、精神紧张(中枢神经反应)。注射腾喜龙无力症状不见好转,反而加重。
(3)反拗性危象 对胆碱酯酶抑制药暂时失效,加大剂量无济于事。
诊断困难病例可采用疲劳(Jolly)试验、腾喜龙或新斯的明试验、AchR-Ab测定、神经重复电刺激检查等。
一、发病原因
1.重症肌无力是一种影响神经肌肉接头传递的自身免疫性疾病,其确切的发病机理目前仍不明确,但是有关该病的研究还是很多的,其中,研究最多的是有关重症肌无力与胸腺的关系,以及乙酰胆碱受体抗体在重症肌无力中的作用,且大量的研究发现,重症肌无力患者神经肌肉接头处突触后膜上的乙酰胆碱受体(AchR)数目减少,受体部位存在抗AchR抗体,且突触后膜上有IgG和C3复合物的沉积。并且证明,血清中的抗AchR 抗体的增高和突触后膜上的沉积所引起的有效的AchR数目的减少,是本病发生的主要原因。而胸腺是AchR抗体产生的主要场所,因此,本病的发生一般与胸腺有密切的关系。所以,调节人体AchR,使之数目增多,化解突触后膜上的沉积,抑制抗AchR抗体的产生是治愈本病的关键。
2.遗传易感性 近年来人类白细胞抗原(HLA)研究显示,MG发病可能与遗传因素有关,根据MG发病年龄、性别、伴发胸腺瘤、AChR-Ab阳性、HLA相关性及治疗反应等综合评定,MG可分为两个亚型:具有HLA-A1、A8、B8、B12和DW3的MG病人多为女性,20~30岁起病,合并胸腺增生,AChR-Ab检出率较低,服用抗胆碱酯酶药疗效差,早期胸腺摘除效果较好;具有HLA-A2、A3的MG病人多为男性,40~50岁发病,多合并胸腺瘤,AChR-Ab检出率较高,皮质类固醇激素疗效好;在国内许贤豪诊治的850例MG病人中,有双胞胎2对(均为姐妹)。
3.近年研究发现,MG与非MHC抗原基因,如T细胞受体(TCR)、免疫球蛋白、细胞因子及凋亡(apoptosis)等基因相关,TCR基因重排不仅与MG相关,且可能与胸腺瘤相关,确定MG病人TCR基因重排方式不仅可为胸腺瘤早期诊断提供帮助,也是MG特异性治疗基础。
4.MG病人外周血单个核细胞(MNC)肾上腺糖皮质激素受体减少,血浆皮质醇水平正常,动物实验提示肾上腺糖皮质激素受体减少易促发EAMG。
二、发病机制
正常疲劳是肌肉连续收缩释放出Ach数量递减,MG的肌无力或肌肉病态疲劳是NMJ处AchR减少导致传递障碍。Ach与AchR结合后产生足以使肌纤维收缩的终板电位,MG的NMJ由于AchR数目减少及抗体竞争作用,使终板电位不能有效地扩大为肌纤维动作电位,运动终板传递受阻使肌肉收缩力减弱,此变化首先反映在运动频率最高、AchR最少的眼肌和脑神经支配肌肉。
用125I标记的α-银环蛇毒素与人类骨胳肌提取的乙酰胆碱受体结合的复合物,可测得病人血清中抗乙酰胆碱受体抗体,抗体阻滞降解突触后膜受体,使自身抗原(nAchR)活性降低,突触后膜表面积减少。由于神经-肌肉接头传递障碍,因而出现肌无力症状。后期肌纤维变性萎缩,纤维组织取而代之。
Fambrough等(1973)证实MG基本缺陷是NMJ突触后膜上AchR明显缺乏,并在EAMG动物血清中检出AchR-Ab,用免疫荧光法可在突触后膜发现AchR与AchR-Ab及补体的免疫复合物沉积。MG患者肌肉活检切片也发现AchR明显减少,从而确定AchR-Ab的致病性,为MG自身免疫学说提供有力证据。将人类AchR-Ab注入正常动物可使之发病,这些证据可满足自身抗体介导性疾病的诊断标准(Drachman)。约85%的全身型MG及50%的眼肌型MG患者可检出AchR-Ab,MG母亲的新生儿也可发现AchR-Ab,使该抗体成为诊断MG敏感可靠的指标。
MG的自身免疫应答异常尚未阐明,约70%的MG病人胸腺异常,其中10%~15%合并胸腺瘤,50%~60%合并胸腺肥大及淋巴滤泡增生,切除胸腺后病情改善。胸腺为免疫中枢,在重症肌无力发病中起重要作用。不论是上皮细胞(包括肌样细胞),还是胸腺(淋巴)细胞遭到免疫攻击,打破免疫耐受性,发生体液免疫(如nAchR-Ab)和细胞免疫(如致敏T细胞),均引起针对nAchR的自体免疫反应,因而发病。胸腺是T细胞成熟场所,在胸腺中已检出AchR亚单位mRNA,MG患者胸腺源性T细胞及B细胞对AchR应答较外周血同类细胞强。此外,正常及增生的胸腺均含有肌样细胞(myoid cells),该细胞类似横纹肌并载有AchR。在某些特定遗传素质个体,某种病毒具有对载有AchR胸腺肌样细胞趋向性(tropism),可损伤细胞并导致细胞表面AchR构型变化,诱导AchR-Ab形成,也有致肿瘤的潜在危险,可能是约10%的MG患者发生胸腺瘤的原因。MG病人胸腺富含AchR致敏T细胞,IgG型AchR-Ab由抗原特异性T辅助细胞(CD4 )激活,由周围淋巴器官、骨髓及胸腺中浆细胞产生。但胸腺不是AchR-Ab惟一来源,胸腺全部摘除后病人仍可长期存在AchR-Ab,可能通过抗原特异性辅助T细胞刺激外周淋巴细胞产生AchR-Ab。
MG病人可有自然杀伤T细胞(nature killer T-cell,NKT)及其他淋巴细胞表型变化,此种NKT细胞数目及功能异常是否与MG发病有关还不清楚。
MG病人常伴其他自身免疫性疾病,如系统性红斑狼疮、风湿性和类风湿性关节炎、干燥综合征、甲状腺功能亢进、甲状腺炎及多发性肌炎等,有些MG病人虽不合并自身免疫病,但可检出自身抗体,如抗甲状腺微粒体及球蛋白抗体、抗核抗体、抗胃壁细胞抗体和抗胰岛B细胞抗体等。
MG患者HLA-B8、DR3和DQB1基因型频率较高,提示发病可能与遗传因素也有关。
主要病理改变如下:
MG骨骼肌改变分为凝血性坏死、淋巴溢及炎性纤维变性三个阶段。8%~20%的MG患者发生肌萎缩,常见神经源性和肌源性损害,可见肌纤维直径大小不一的断裂、增殖、核向中央移位、玻璃样变性和结缔组织增生等。
青少年患者肌肉损害发生率较高,约42%,儿童仅为12%。最重要病变发生在运动终板超微结构水平,Engel等(1976)电镜观察本病神经末梢及面积减少,NMJ突触前膜变宽,囊泡数量及所含ACh量为正常范围。突触后膜延长,初级突触间隙由正常的200A°增宽至400~600A°,突触皱褶减少、变浅,表面破碎和皱缩,缺乏次级皱褶,突触间隙可见基底膜样物质聚积,构成神经肌肉传导阻滞基础,称为突触间失神经作用。
约60%的MG患者发生胸腺淋巴样增生(lymph oid hyperplasia),局限于胸腺髓质生发中心,年轻病人出现率高。10%~15%的MG病人合并胸腺瘤,MG合并胸腺瘤病理组织学改变可分为三型:上皮细胞型、淋巴细胞型及混合细胞型(上皮细胞与淋巴细胞),少见胸腺瘤有梭形细胞瘤及霍奇金肉芽肿,各占约1%。胸腺瘤一般为良性。
常见并发症:重症肌无力危象、类风湿性关节炎、红斑狼疮、溶血性贫血
由于肌无力病者因呼吸、吞咽困难而不能维持基本生活、生命体征时,称为肌无力危象。发生率约占肌无力总数的9.8%~26.7%。
根据肌无力危象发生的原因可分为3种类型:肌无力性危象、胆碱能性危象和反拗性危象。
1.肌无力性危象系由疾病发展和抗胆碱酯酶药物不足所引起。临床表现吞咽、咳嗽不能,呼吸窘迫、困难乃至停止的严重状况。体检可见瞳孔扩大,浑身出汗、腹胀、肠鸣音正常和新斯的明注射后症状好转等特点。
2.胆碱能性危象约占危象例数的1.0%~6.0%,由于抗胆碱酯酶过量所引起。除肌无力的共同特点外,病者瞳孔缩小,浑身出汗,肌肉跳动,肠鸣音亢进,肌注新斯的明后症状加重等特征。
3.反拗性危象是由感染、中毒和电解质紊乱所引起,应用抗胆碱酯酶药物后可暂时减轻,继之又加重的临界状态。
重症肌无力可伴有其他疾病,如胸腺瘤、其次为甲状腺功能亢进,少数可伴有类风湿关节炎、红斑狼疮和自体溶血性贫血等。
最近浏览

