发布时间:2020年03月31日 13:14:11 来源:振东健康网
张展 张岱 肖冰冰 张瑞 白会会 董翰宇 毕蕙 刘朝晖
【摘 要】目的了解高危型HPV感染妇女阴道菌群及子宫颈菌群的情况,探讨高危型HPV感染与阴道菌群及子宫颈菌群的关系。方法选取2017年9月至10月北京大学第一医院妇产科就诊的患者(对照组5例,HPV16/18组5例,HPV其他高危型组5例,子宫颈癌组3例),填写调查问卷、行阴道及子宫颈分泌物微生态检测,同时使用新一代测序技术扩增细菌16SrRNA基因V3~V4高变区行菌群分析。结果阴道菌群分析结果:(1)阴道菌群主要有6个菌门:厚壁菌门、放线菌门、拟杆菌门、变形菌门、梭菌门及软壁菌门,正常阴道菌群以厚壁菌门为主,高危型HPV感染者拟杆菌门、梭菌门增多,子宫颈癌组梭菌门显著增多;(2)正常阴道菌群在属水平以乳杆菌属为主,高危型HPV感染者乳杆菌减少,加德纳菌属、普雷沃菌属、阿托波菌属、巨球菌属及纤毛菌属增多,子宫颈癌组纤毛菌属显著增多;(3)高危型HPV感染后阴道菌群多样性增加,但4组阴道菌群Alpha多样性无显著性差异(P=0.073)。子宫颈菌群分析结果:(1)子宫颈菌群多样性高于阴道菌群;(2)高危型HPV感染者子宫颈菌群多样性明显增加,4组子宫颈菌群Alpha多样性有显著性差异(P=0.046);(3)对照组子宫颈菌群较阴道菌群变形菌门增加,HPV感染后子宫颈变形菌门明显增加;(4)衣原体感染在子宫颈癌组明显增加。结论子宫颈菌群多样性高于阴道菌群;高危型HPV感染者子宫颈菌群变化较阴道菌群更明显;梭菌门-纤毛菌属及衣原体与子宫颈癌相关,子宫颈变形菌门可能与高危型HPV感染相关。
【关键词】乳头状瘤病毒感染;微生物群;阴道;子宫颈;高通量核苷酸系列分析
Primary study on the relationship between high-risk HPV infection and vaginal cervical microbiota
Zhang Zhan, Zhang Dai, Xiao Bingbing, Zhang Rui, Bai Huihui, Dong Hanyu, Bi Hui, Liu Zhaohui. Department of Obstetrics and Gynecology, Peking University First Hospital, Beijing 100034, China
Corresponding author: Liu Zhaohui
【Abstract】 Objective To understand characteristics of vaginal cervical microbiota in high-risk
HPV(hrHPV)infected women and to uncover the relationship between hrHPV infection and vaginalcervical microbiota. Methods All participants were randomly selected from Peking University First.Hospital from September to October of 2017, including 5 subjects of control group, 5 cases of HPV16/18 group, 5 cases of other hrHPV infected group and 3 cases of cervical squamous carcinoma group. All subjects were required to fill in a questionnaire, and cervical and vaginal discharges were separately collected for microscopic examination and new generation sequencing targeting the variable region(V3-V4)of bacterial 16S rRNA gene. Results Vaginal microbiota analysis:(1)6 major phylum were found in vaginal microbiota:Firmicutes, Bacteroidetes, Fusobacteria, Actinobacteria, Tenericutes and Proteobacteria.Firmicutes contributed to the majority of normal vaginal flora, Bacteroidetes and Fusobacteria increased in hrHPV infected ones, while Fusobacteria showed significant difference in cervical carcinoma group.(2)Lactobacillus occupied most of normal vaginal flora while genus like Gardnella, Prevotella, Atopobium, Megasphaera and Sneathia increased in hrHPV infected subjects, Sneathia showed significant difference in cervical carcinoma group.(3)No significant difference had been calculated in Alpha diversity of four.groups(P=0.073). Cervical microbiota analysis:(1)Microbial diversity of cervical microbiota was higher than that of vaginal microbiota.(2)Significant difference had been found in Alpha diversity of four groups(P=0.046).(3) Proteobacteria in normal cervical flora was much more than that in vagina, and Proteobacteria increased significantly in hrHPV infected cervical discharge. (3) Chlamydia increased significantly in cervical carcinoma group. Conclusions The diversity of cervical microbiota is higher than that of vaginal microbiota. Change in cervical microbiota is more obvious than that of vagina in hrHPV infected subjects. Fusobacteria-Sneathia and Chlamydia significantly increase in cervical carcinoma group. Proteobacteria might relate to hrHPV infection.
【Key words】 Papillomavirus infections; Microbiota; Vagina; Cervix uteri; High-throughput;nucleotide sequencing
高危型 HPV(high-risk, HPV,hrHPV)持续感染与子宫颈癌的发生密切相关[1-3]。70%的子宫颈癌是由HPV16、18型持续感染所致[4]。随着新一代测序(new generation sequencing,NGS)技术的发展,越来越多的研究证据提示,下生殖道的菌群环境可能与hrHPV感染、感染的持续、黏膜局部免疫及子宫颈病变有关[5]。目前,对下生殖道菌群的研究多集中于阴道菌群(vaginal microbiota),认为不同类型阴道菌群可能影响 HPV 感染的清除、持续或子宫颈病变的发生。本研究关注hrHPV感染者子宫颈菌群(cervical microbiota)的特点,并将其与阴道菌群进行对比,以深入研究hrHPV感染与下生殖道菌群环境的关系,为揭示hrHPV持续感染及其引起子宫颈病变的机制提供线索。
资料与方法
一、实验分组及标本收集
1. 实验分组:随机选取2017年9—10月就诊于北京大学第一医院妇产科的患者,分为HPV16/18
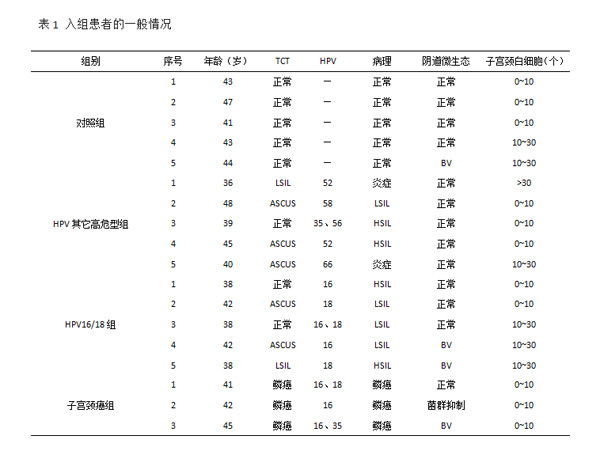
组:HPV16 或(和)18 型(HPV16/18)感染者,5 例;HPV 其他高危型组:除16、18型,HPV 其他高危型感染者,5例;对照组:液基薄层细胞学检查(TCT)及HPV均阴性者,5例,选自因子宫肌瘤行子宫切除术的患者;子宫颈癌组:子宫颈鳞癌者,3例。应用凯普 HPV21 分型检测法,hrHPV 感染指 HPV16、18、31、33、35、39、45、51、52、53、56、58、59、66、68中任一阳性。所有入组者均知情同意,填写调查问卷。入组患者的纳入标准:年龄≥18岁,月经规律,卵泡中期,未绝经,有性生活史,体质指数正常(18.5~23.9 kg/m2),初次发现hrHPV 感染;排除标准:妊娠期、哺乳期或绝经期妇女,无性生活史,长期口服激素或免疫抑制剂者,7 d内应用抗生素、有性生活或进行阴道冲洗者,其他严重的系统疾病或肿瘤。各组在职业、卫生习惯、婚育情况及既往下生殖道感染情况上均无差异(P>0.05)。见表1。
2. 标本收集:(1)分泌物涂片:无菌棉拭子分别采集阴道侧壁上1/3的分泌物及子宫颈分泌物。采集子宫颈分泌物前,先用无菌棉拭子将子宫颈表面的黏液及附着物拭去,再将新的无菌棉拭子伸入颈管,顺时针旋转3~5周。涂片,革兰染色,100×油镜阅片。(2)用于菌群测定的分泌物:无菌棉拭子采集阴道及子宫颈分泌物(方法同上),分别置入含无菌生理盐水的2 ml EP管内,-80 ℃保存,用于菌群测定。
二、菌群测定及分析
本研究采用目前性价比最高的NGS平台IlluminaMiseq平台(美国Illumina公司产品),选取覆盖序列最广且测序质量最高的细菌16SrRNA基因V3~V4高变区进行测序及微生物分类分析。具体步骤如下:
1.细菌 DNA 的提取:参照美国 Omega Bio-Tek公司的 DNA 提取试剂盒 E.Z.N.ATM Mag-Bind Soil
DNA Kit 进行 DNA 提取,琼脂糖凝胶检测 DNA的完整性和纯度。PCR扩增细菌16S rRNA基因V3~V4区,各样本扩增引物前添加特异性标签序列区分;PCR引物融合Illumina Miseq平台的V3~V4通用引物。采用Illumina Miseq平台进行测序。
2.数据处理:去除原始测序的引物接头序列,将重叠的序列进行拼接,按照标签序列区分不同样
本的数据,然后对各样本数据进行质控,去除嵌合体及非特异性扩增序列,得到有效序列。使用Usearch软件对有效序列按照97%相似度分成不同操作分类单元(operational taxonomic units,OTU),选择丰度最高的序列作为 OTU 代表序列,比对rRNA 基因数据库——RDP 数据库(http://rdp.cme.msu.edu/index.jsp)进行物种分类及生物信息学统计分析。
3.分析的内容:(1)OTU聚类分析:各组间OTU有一定的重叠,应用 R 软件绘制韦恩图以直观体
现。(2)Alpha多样性分析:计算衡量样本物种多样性的 Shannon 指数;Alpha 多样性指样本内或组内的物种多样性,以Shannon指数表示Alpha多样性,数值越高,则菌群多样性越高。(3)物种分类分析:将OTU按照RDP数据库进行6个水平的物种分类,即界(domain)、门(phylum)、纲(class)、目(order)、科(family)、属(genus);计算各物种的丰度,并绘制统计图。(4)菌群差异分析:应用线性判别分析
(linear discriminant analysis effect size,LEfSe)寻找组间或样本间的菌群差异。
三、数据分析及统计学方法
数据预处理及OTU聚类时主要应用Cutadapt、Usearch、prinseq-lite、pear 及 uchime 软件,Alpha 多样性分析主要应用Mothur软件及方差分析,物种分类分析主要应用 RDPclassifier 软件,LEfSe 主要应用 LEfSe 软件、Kruskal-Wallis 秩和检验、Wilcoxon秩和检验,患者一般资料的分析包括方差分析、Fisher精确概率法检验等。
结 果
一、OTU聚类分析
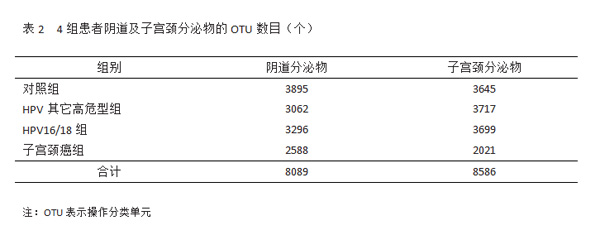
通过NGS测序,4组患者阴道及子宫颈分泌物分别获得8089及8586个OTU,见表2。各组OTU
有重叠,见图 1。hrHPV 感染者(即 HPV16/18 组、HPV其他高危型组)子宫颈菌群OTU数目更多,菌
群更丰富,见表2。
二、Alpha多样性分析
使用 Shannon 指数计算 Alpha 多样性,数值越高,则菌群多样性越高,结果显示,对照组、hrHPV组及子宫颈癌组阴道及子宫颈菌群多样性逐渐升高。4 组的子宫颈菌群多样性有显著性差异(P=0.046)。但4组阴道菌群Alpha多样性无显著性差异(P=0.073)。各组阴道菌群与子宫颈菌群分别比较,HPV16/18组的子宫颈菌群多样性明显高于阴道菌群(P=0.034),其余 3 组阴道与子宫颈菌群多样性无显著性差异(P>0.05)。
三、物种分类分析
OTU按照RDP数据库进行6个水平的物种分类,分析得出,阴道分泌物在门水平主要有厚壁菌门(Firmicutes)、放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、梭菌门(Fusobacteria)、变形菌门(Proteobacteria)及软壁菌门(Tenericutes)。
阴道菌群分析结果:正常阴道菌群以厚壁菌门为主,hrHPV感染者拟杆菌门增多,HPV16/18感染及子宫颈癌者厚壁菌门减少,梭菌门增加;子宫颈癌组梭菌门显著增多。正常阴道菌群在属水平以乳杆菌属(Lactobacillus)为主,hrHPV 感染者乳杆菌减少,加德纳菌属(Gardnella vaginalis)、阿托波菌属(Atopobium)、普雷沃菌属(Prevotella)、巨球菌属(Megasphaera)增多,HPV16/18 感染及子宫颈癌者纤毛菌属(Sneathia)明显增多。子宫颈菌群分析结果:正常子宫颈菌群与阴道菌群相比,变形菌门明显增加。对照组及子宫颈癌组阴道菌群与子宫颈菌群多样性差异不大,而hrHPV感染者子宫颈菌群多样性明显高于阴道菌群,见图2。HPV16/18感染者及子宫颈癌者子宫颈分泌物中梭菌门-纤毛菌属及衣原体检出明显增加。
在此基础上,本研究将hrHPV感染者(即HPV16/18组、HPV其他高危型组)TCT及子宫颈病理结果分别进一步分组,分析hrHPV感染状态下不同子宫颈病变程度患者阴道及子宫颈菌群的变化,
表5及表6显示了两组中不同TCT或病理结果患者阴道及子宫颈6个菌门所占的构成比;结果显示,无论是按照TCT或是子宫颈病理结果分组,阴道及子宫颈 6 个菌门均未随着子宫颈病变程度的加重
表现出规律性的变化。相较于子宫颈病变程度,下生殖道菌群与hrHPV感染的关系可能更为密切。
四、菌群差异分析
应用 LEfSe 分别对比各组的阴道菌群与子宫颈菌群,结果显示,(1)hrHPV组的子宫颈菌群较阴
道菌群差异较大,以变形菌门增加最为显著,变形菌门可能与hrHPV感染相关;(2)对照组
子宫颈菌群较阴道菌群有差异,变形菌门、芽孢杆菌目(Bacillales)及链球菌科(Streptococcaceae)-乳球菌属(Lactococcus)增多;(3)子宫颈癌组的子宫颈菌群和阴道菌群均极其复杂,两者无显著性差异
讨 论
一、阴道菌群的特点及其与hrHPV感染的关系
女性下生殖道是个复杂、独特且动态变化的微生态体系。微生态及菌群是下生殖道微生态研究的核心内容[6]。随着分子生物学技术的发展,NGS技术即二代高通量测序技术使阴道菌群的研究取得了飞速突破。Ravel等[7]在2011年首次提出将阴道菌群分为5个类群(community state types,CST),CSTⅠ、Ⅱ、Ⅲ、Ⅴ分别代表了以卷曲乳杆菌、加氏乳杆菌、惰性乳杆菌及詹氏乳杆菌为优势的类型,CST Ⅳ以厌氧菌为优势菌群、乳杆菌明显减少,Ⅳ-A以厌氧球菌、噬冻菌属及普雷沃菌属等为主,Ⅳ-B 型以阿托波菌及巨球菌属等为主。CSTⅠ、Ⅱ、Ⅴ型代表正常的阴道菌群类型,CST Ⅳ型代表多样性明显增加的异常阴道菌群类型,而以惰性乳杆菌为代表的CST Ⅲ型则代表了阴道菌群的亚健康状态,由于惰性乳杆菌不能产生 H2O2以维持阴道的低pH环境[8-10],致使该类菌群最容易向Ⅳ型转化并诱发疾病[11]。
阴道菌群受多种因素的影响,如种族、激素水平、吸烟、性交及阴道冲洗等卫生习惯。研究发现,黑人女性更容易出现阴道菌群多样性增加及菌群图异常,这可能与基因及卫生习惯差异有关[7]。吸烟、性交、阴道冲洗等均可能不同程度造成阴道菌群多样性增加、乳杆菌下降[12-14]。激素水平对阴道菌群的影响则体现在女性一生的不同时期,表现为高雌激素状态有利于阴道菌群向乳杆菌优势转化并维持稳定[15-18]。本研究选取月经规律、卵泡中期、近期无性生活且未应用抗生素的育龄期妇女,入组者在年龄、生活、卫生习惯上无明显差异,最大程度避免了混杂因素对菌群的影响。
关于阴道微生物致病性的研究最早可以追溯到20余年前,Platz-Christensen等[19]发现,以加德纳菌等厌氧菌为优势的细菌性阴道病可能在子宫颈癌癌前病变的发生中发挥重要作用。随后,Smith等[20]发现,沙眼衣原体感染可能增加子宫颈鳞癌的发生风险。而生殖道菌群与导致子宫颈病变的HPV 感染之间的关系也在近年来逐渐明确。Lee等[21]在2013年首次应用NGS技术分析了HPV感染与阴道菌群的关系,认为 HPV 阳性者阴道菌群多样性增加同时乳杆菌属减少。这可能缘于大部分乳杆菌可以产生乳酸及 H2O2维持阴道的低 pH 环境[22-24],同时可以产生并分泌细菌素以抑制其他厌氧菌(如加德纳菌)黏附及生物膜的形成[25-26]。在此基础上,Brotman等[27]对32例性活跃的育龄期妇女进行了长达 16 周的阴道分泌物自采样研究,显示阴道菌群类型与 HPV 的持续或清除相关,CST Ⅳ型菌群者更容易感染 HPV 且清除较慢,而加氏乳杆菌优势型阴道菌群者清除HPV较快。本研究通过分析4组患者的阴道菌群发现,阴道菌群在门水平主要有6个菌门,包括厚壁菌门、放线菌门、拟杆菌门、梭菌门、变形菌门及软壁菌门。正常人阴道菌群以厚壁菌门为主,hrHPV组厚壁菌门减少、拟杆菌门及梭菌门增加,菌群多样性增加。阴道梭菌门-纤毛菌属及衣原体在子宫颈癌组明显增加,提示可能与 hrHPV 感染甚至子宫颈癌变相关,梭菌门-纤毛菌属可能为hrHPV感染导致子宫颈癌的目标菌种;既往研究也曾观察到,扫描电镜下梭菌门-纤毛菌属对于子宫颈癌细胞的黏附及细胞膜破坏作用[28]。可以推测,细胞膜的破坏有利于HPV侵入鳞状上皮细胞及进一步的基因整合[29-30],促进了子宫颈癌的发生。此外,Alpha多样性分析提示,4组患者阴道菌群多样性虽有差异但并无显著性差异,可能与样本例数偏少有关。
关于阴道菌群与子宫颈病变的关系,既往曾有4个较为典型的横断面研究,但结果一直存在争议。Mitra 等[31]、Audirac-Chalifour 等[32]及 Oh 等[33]认为,子宫颈病变与阴道菌群多样性增加及乳杆菌减少有关,随着子宫颈病变程度的升高,CST Ⅳ型菌群明显增加。然而,Piyathilake 等[34]应用 Dirichlet多项混合模型研究了340例高级别鳞状上皮内病变(HSIL)妇女及 90 例低级别鳞状上皮内病变(LSIL)妇女的阴道分泌物,却并未发现阴道菌群多样性增加与HSIL的相关性。本研究也未发现子宫颈病变程度与阴道菌群、子宫颈菌群的相关性。这一矛盾的结果可能是由于横断面研究及样本例数的限制,因此,更大规模更长时间的前瞻性研究有待进行。
二、子宫颈菌群与hrHPV感染的关系
1.阴道菌群与子宫颈菌群:由于“女性上生殖道无菌”这一传统观念的限制,既往有关女性生殖道菌群的研究大都集中于阴道菌群。然而,2017年,我国学者吴瑞芳首次发现女性子宫颈及上生殖道同样分布着众多菌群,且女性生殖道自下而上存在着菌群类型的差异及一定的相互转化关系[35]。菌群由下生殖道的厚壁菌门优势类型逐渐演变为上生殖道的变形菌门、放线菌门及拟杆菌门等优势类型。子宫颈菌群相比于阴道菌群表现为乳杆菌属减少,菌群多样性增加。本研究中对照组子宫颈菌群与阴道菌群相比,变形菌门、芽孢杆菌目及链球菌科明显增加,与前述研究结果一致。
2.子宫颈菌群与 hrHPV 感染:在阴道菌群与HPV感染关系逐渐明确之后,可以推测hrHPV感染
者的子宫颈菌群可能同样有别于阴道菌群。本研究分析发现,子宫颈菌群的多样性高于阴道菌群,且4组患者子宫颈菌群多样性有显著性差异,表现为从对照组到hrHPV组到子宫颈癌组,菌群多样性逐渐增加。进一步对比各组患者的阴道菌群与子宫颈菌群发现,对照组子宫颈菌群较阴道菌群更丰富,以变形菌门为主的少部分菌增加。子宫颈癌组子宫颈菌群及阴道菌群均呈现出极其复杂的多样性,两者无显著性差异,这可能与子宫颈癌患者阴道不规则出血造成阴道及子宫颈分泌物混合有关。hrHPV组尤其是HPV16/18组子宫颈菌群多样性明显高于阴道菌群。hrHPV感染者阴道微生态正常的情况下,子宫颈菌群已经出现巨大变化。对hrHPV组阴道菌群与子宫颈菌群进行LEfSe分析可见,hrHPV组子宫颈菌群变形菌门大幅增加。由于正常阴道内缺乏变形菌门,而既往研究又较少涉及HPV感染状态下的子宫颈菌群,这一新发现可能预示变形菌门与hrHPV感染相关。
由此认为,女性下生殖道菌群与hrHPV感染有密切关系。子宫颈菌群作为下生殖道菌群的重要组成部分,有自身的特殊性,不应被忽略。本研究在以往研究的基础上,将hrHPV感染者子宫颈菌群与阴道菌群进行对比,进一步揭示了下生殖道菌群与hrHPV感染的关系。未来应扩大样本量,以求准确分析子宫颈菌群对hrHPV感染及子宫颈病变的潜在影响,寻找目标致病菌,揭示hrHPV感染状态下菌群与局部免疫的关系及相关机制;设计高质量的前瞻性研究分析HPV持续感染人群的生殖道菌群情况。相信未来应用菌群预测、预防甚至治疗下生殖道感染性疾病将会成为可能。
参考文献
[1]Moody CA, Laimins LA. Human papillomavirus oncoproteins:pathways to transformation[J]. Nat Rev Cancer, 2010, 10(8):550-560. DOI: 10.1038/nrc2886.
[2] oscicki AB. Human papilloma virus, papanicolaou smears,and the college female[J].
Pediatr Clin North Am, 2005, 52(1):163-177, ix. DOI: 10.1016/j.pcl.2004.10.005.
[3]Plummer M, Schiffman M, Castle PE, et al. A 2-year prospective study of human papillomavirus persistence among women with a cytological diagnosis of atypical squamous cells of undetermined significance or low-grade squamous intraepithelial lesion[J]. J Infect Dis, 2007, 195(11):1582-1589. DOI: 10.1086/516784.
[4]Muñoz N, Bosch FX, Castellsagué X, et al. Against which human papillomavirus types shallwe vaccinate and screen?The international perspective[J]. Int J Cancer, 2004, 111(2):278-285. DOI: 10.1002/ijc.20244.
[5]Mitra A, MacIntyre DA, Marchesi JR, et al. The vaginal microbiota, human papillomavirus infection and cervical intraepithelial neoplasia: what do we know and where are we going next? [J]. Microbiome, 2016, 4(1): 58. DOI: 10.1186 /s40168-016-0203-0.
[6]肖冰冰. 阴道微生态的分子生物学研究[J]. 中国实用妇科与产科杂志,2017,33(8):795-800.
[7]Ravel J, Gajer P, Abdo Z, et al. Vaginal microbiome of reproductive-age women[J]. Proc Natl Acad Sci U S A, 2011,108 Suppl 1:4680-4687. DOI: 10.1073/pnas.1002611107.
[8]Klebanoff SJ, Coombs RW. Viricidal effect of Lactobacillus acidophilus on human immunodeficiency virus type 1:possible role in heterosexual transmission[J]. J Exp Med,1991, 174(1):289-292.
[9]Klebanoff SJ, Hillier SL, Eschenbach DA, et al. Control of the microbial flora of the vagina by H2O2-generating lactobacilli[J]. J Infect Dis, 1991, 164(1):94-100.
[10]Clark RA, Klebanoff SJ. Role of the myeloperoxidase-H2O2-halide system in concanavalin A-induced tumor cell killing by human neutrophils[J]. J Immunol, 1979, 122(6):2605-2610.
[11]Romero R, Hassan SS, Gajer P, et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women[J].Microbiome,2014, 2(1):4. DOI: 10.1186/2049-2618-2-4.
[12]Brotman RM, He X, Gajer P, et al. Association between cigarette smoking and the vaginal microbiota: a pilot study[J].BMC Infect Dis, 2014, 14:471. DOI: 10.1186/1471-2334-14-471.
[13]Mändar R, Punab M, Borovkova N,et al. Complementary seminovaginal microbiome in couples[J]. Res Microbiol, 2015,166(5):440-447. DOI: 10.1016/j.resmic.2015.03.009.
[14]Schwebke JR, Desmond RA, Oh MK. Predictors of bacterial vaginosis in adolescent women who douche[J]. Sex TransmDis, 2004, 31(7):433-436.
[15]Hickey RJ, Zhou X, Settles ML, et al. Vaginal microbiota of adolescent girls prior to the onset of menarche resemble those of reproductive-age women[J]. MBio, 2015, 6(2): pii:e00097-15. DOI: 10.1128/mBio.00097-15.
[16]Gajer P, Brotman RM, Bai G, et al. Temporal dynamics of the human vaginal microbiota[J]. Sci Transl Med, 2012, 4(132):132ra52. DOI: 10.1126/scitranslmed.3003605.
[17]Brotman RM, Shardell MD, Gajer P, et al. Association between the vaginal microbiota, menopause status, and signs of vulvovaginal atrophy[J]. Menopause, 2014, 21(5): 450-458.
DOI: 10.1097/GME.0b013e3182a4690b.
[18]MacIntyre DA, Chandiramani M, Lee YS, et al. The vaginal microbiome during pregnancy and the postpartum period in a European population[J]. Sci Rep, 2015, 5:8988. DOI: 10.1038/srep08988.
[19]Platz-Christensen JJ, Sundström E, Larsson PG. Bacterial vaginosis and cervical intraepithelial neoplasia[J]. Acta Obstet Gynecol Scand, 1994, 73(7):586-588
[20]Smith JS, Bosetti C, Muñoz N, et al. Chlamydia trachomatis and invasive cervical cancer: a pooled analysis of the IARC multicentric case-control study[J]. Int J Cancer, 2004, 111(3):431-439. DOI: 10.1002/ijc.20257.
[21]Lee JE, Lee S, Lee H, et al. Association of the vaginal microbiota with human papillomavirus infection in a Korean twin cohort[J].PLoS One, 2013, 8(5): e63514.DOI:10.1371/journal.pone.0063514.
[22]Mastromarino P, Di Pietro M, Schiavoni G, et al. Effects of vaginal lactobacilli in Chlamydia trachomatis infection[J].Int J Med Microbiol,2014,304(5-6): 654-661.DOI:10.1016/j.ijmm.2014.04.006.
[23]Breshears LM, Edwards VL, Ravel J, et al. Lactobacillus crispatus inhibits growth of Gardnerella vaginalis and Neisseria gonorrhoeae on a porcine vaginal mucosa model[J].BMC Microbiol, 2015, 15:276. DOI: 10.1186/s12866-015-0608-0.
[24]Graver MA, Wade JJ. The role of acidification in the inhibition of Neisseria gonorrhoeae by vaginal lactobacilli during anaerobic growth[J]. Ann Clin Microbiol Antimicrob, 2011, 10:8. DOI: 10.1186/1476-0711-10-8.
[25]Reid G, Heinemann C, Velraeds M, et al. Biosurfactants produced by Lactobacillus[J]. Methods Enzymol, 1999, 310:426-433.
[26]Ojala T, Kankainen M, Castro J, et al. Comparative genomics of Lactobacillus crispatus suggests novel mechanisms for the competitive exclusion of Gardnerella vaginalis[J]. BMC
Genomics, 2014, 15:1070. DOI: 10.1186/1471-2164-15-1070.
[27]Brotman RM, Shardell MD, Gajer P, et al. Interplay between the temporal dynamics of the vaginal microbiota and human papillomavirus detection[J]. J Infect Dis, 2014, 210(11):1723-1733. DOI: 10.1093/infdis/jiu330.
[28]Harwich MD Jr, Serrano MG, Fettweis JM, et al. Genomic sequence analysis and characterization of Sneathia amnii sp.nov[J]. BMC Genomics, 2012, 13 Suppl 8: S4. DOI: 10.1186 /1471-2164-13-S8-S4.
[29]Briselden AM, Moncla BJ, Stevens CE, et al. Sialidases (neuraminidases) in bacterial vaginosis and bacterial vaginosis-associated microflora[J]. J Clin Microbiol, 1992, 30(3):663-666.
[30]Holmes KK, Chen KC, Lipinski CM, et al. Vaginal redox potential in bacterial vaginosis (nonspecific vaginitis) [J]. J Infect Dis, 1985, 152(2):379-382.
[31]Mitra A, MacIntyre DA, Lee YS, et al. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity[J]. Sci Rep, 2015, 5:16865. DOI:10.1038/srep16865.
[32]Audirac-Chalifour A, Torres-Poveda K, Bahena-Román M, et al. Cervical Microbiome and Cytokine Profile at Various Stages of Cervical Cancer: A Pilot Study[J]. PLoS One, 2016,11(4):e0153274. DOI: 10.1371/journal.pone.0153274.
[33]Oh HY, Kim BS, Seo SS, et al. The association of uterine cervical microbiota with an increased risk for cervical intraepithelial neoplasia in Korea[J]. Clin Microbiol Infect,2015, 21(7):674.e1-9. DOI: 10.1016/j.cmi.2015.02.026.
[34]Piyathilake CJ, Ollberding NJ, Kumar R, et al. Cervical Microbiota Associated with Higher Grade Cervical Intraepithelial Neoplasia in Women Infected with High-Risk Human Papillomaviruses[J]. Cancer Prev Res (Phila), 2016, 9(5):357-366. DOI:10.1158/1940-6207.CAPR-15-0350.
[35]Chen C, Song X, Wei W, et al. The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases[J]. Nat Commun, 2017, 8(1): 875. DOI: 10.1038/s41467-017-00901-0.


移动平台
微信平台
