发布时间:2020年11月02日 09:02:31 来源:振东健康网
Yang Yang, 1 Mayu Sun, 1 Wenbo Yao, 1 Feng Wang, 1 Xiaoguang Li, 2 Wei Wang, 3Jingquan Li, 2 Zhihu Gao, 1 Lin Qiu, 1 Rongli You, 3 Chenghua Yang, 1 Qian Ba, 2Hui Wang
1中国科学院上海生命科学研究院营养与健康研究所,营养代谢与食品安全重点实验室
2上海交通大学医学院公共卫生学院,单细胞组学与疾病研究中心,癌基因及相关基因国家重点实验室
3北京振东光明药物研究院有限公司药理部
【摘要】
背景:目前,肝细胞癌(HCC)仍缺乏有效的治疗方法。免疫治疗联用传统的治疗手段为肝细胞癌的治疗带来新的希望。本研究旨在探究中药复方苦参注射液(CKI)的免疫调节作用及其与低剂量索拉菲尼联合抗肝癌作用。
方法:构建两种肝癌细胞系的小鼠原位肿瘤模型,皮下肿瘤模型,两种术后复发模型,肿瘤切除后再移植模型来观察肿瘤的原位生长情况,复发率以及免疫记忆效果;中和抗体分别敲除小鼠体内巨噬细胞以及CD8+ T细胞后观察药物治疗效果;体外实验,利用小鼠来源的原代巨噬细胞以及CD8+ T细胞来探究复方苦参注射液对相关免疫细胞的具体影响。
结论:复方苦参注射液显著地增强了低剂量索拉菲尼的治疗肝癌的效果,并具有良好的生物安全性。复方苦参注射液联用低剂量索拉菲尼预防了肝癌的术后复发以及抑制了再种植的肿瘤生长。进一步的研究发现,复方苦参注射液通过激活巨噬细胞表面TNFR1介导的NF-κB和p38 MAPK信号通路,促进肿瘤相关巨噬细胞(TAM)向促炎性M1-TAM分化,同时抑制抑炎M2-TAM分化。复方苦参注射液刺激后的巨噬细胞提高了肿瘤微环境中CD8+ T细胞的增殖能力及杀伤功能,同时降低了CD8+ T细胞功能耗竭,最终导致肝癌细胞的凋亡。
总结:复方苦参注射液通过影响巨噬细胞以及CD8+ T细胞,重塑了肝癌的肿瘤免疫微环境,增敏了低剂量索拉菲尼抗肝癌的治疗效果,同时避免了化疗药物引起的毒副作用。我们的研究结果表明具有免疫调节功能的传统中药复方增敏化疗药物的治疗效果,复方苦参注射液可作为肝癌治疗的一种候选药物。
【介绍】
肝细胞癌(Hepatocellular carcinoma,HCC)是最常见的原发性恶性肝癌,发病率约占全球总发病率的第六位,致死率位居第四位1 2。肝细胞癌的发病率在亚洲东部、南部和中部最高。3。目前治疗肝癌的方法包括经动脉化疗栓塞术(TACE),外科手术切除、放疗和化疗4。虽然肝切除和肝移植仍然是早期肝癌患者最有效的治疗方法,但它们不适用于大多数在确诊时就已处于晚期肝癌的患者。对于晚期肝癌患者来说,FDA仅批准了3个用于临床治疗的化疗药物,包括一线疗法索拉菲尼和乐伐替尼,二线疗法瑞格菲尼。由于缺乏更有效的治疗方法,肝癌是第二致命的癌症类型,5年生存率仅为18%2。因此,迫切需要开发新的肝癌治疗策略,特别是前景良好的联合治疗,以最大限度地提高临床效益,减少毒副作用2。
肝癌肿瘤免疫微环境中的免疫细胞在肿瘤的发生、发展以及转移中发挥了重要的作用。。肿瘤相关巨噬细胞(TAMs)和CD8+T细胞是这些免疫细胞的主要成分9 10。巨噬细胞在肝癌的发生发展中起着关键作用10。通常来说,巨噬细胞可以响应各种体内的信号传导而极化为M1或M2状态。11。经典激活型巨噬细胞,称为M1(由LPS或IFN-γ激活),通过产生促炎型免疫细胞因子(IL-1β,TNFα,INF-β1等),参与抗原呈递并由此产生相应的抗肿瘤活性。替代激活型巨噬细胞,称为M2(IL-4或IL-13激活),产生抗炎型免疫抑制效应子(Arg-1,IL-10,CD206等),发挥促肿瘤作用。在肝癌免疫微环境中,肿瘤细胞可以驯化浸润的巨噬细胞并将其极化为M2-TAM,同时抑制M1-TAM极化,从而进一步导致肿瘤组织中细胞毒性T细胞(CD8+ T)的减少和杀伤功能衰竭。因此,将TAM转化为促炎性M1型巨噬细胞是治疗肝细胞癌的潜在策略。
传统中药治疗疾病已有2500年的历史。复方苦参注射液(CKI)是中国国家药品监督管理局(NMPA)批准的中药配方。CKI是从苦参和白土苓两种药草的根部提取出的。CKI中的主要生物活性生物碱包括氧化苦参碱、苦参碱、氧化槐果碱和槐果碱。在过去的20年中,CKI已在临床上用于治疗各种实体瘤类型,包括肝癌,肺癌,乳腺癌,胃癌,结肠直肠癌和其他癌症类型。值得关注的是,研究发现CKI通过TRPV1信号传导可有效治疗癌症引起的疼痛。先前的报道表明,CKI单独使用或与常规化疗结合使用可以增强抗肿瘤功效,降低化疗诱导的毒性并改善病人的生活质量。对于肝细胞癌的治疗,CKI可提高肝动脉插管化疗栓塞法治疗不可切除的肝癌的治疗效果。但是,关于CKI对肿瘤免疫微环境的调节及其潜在机制的了解仍知之甚少。特别是,考虑到免疫治疗(靶向微环境)和化学治疗药物(靶向肿瘤细胞)的不同作用方式,因此有必要探究CKI和化学治疗药物联合治疗肝细胞癌的潜在功效。
在本研究中,我们将CKI与低于临床剂量的索拉菲尼联合使用来治疗肝细胞癌。 低剂量的选择可降低索拉菲尼治疗给带来的副作用,但与CKI联合治疗后,CKI增强了低剂量索拉菲尼抗癌活性增强。通过调节TAM和T细胞,CKI还减少了肿瘤复发并引发了长期的抗肿瘤记忆反应。进一步的机制研究发现,CKI通过巨噬细胞表面的TNFR1受体以及其下游的NF-κB和p38 MAPK信号通路发挥免疫调节作用。本研究为CKI与低剂量索拉菲尼联合用于肝细胞癌治疗的潜在临床应用提供了证据。
材料与方法
动物模型
从上海斯莱克公司(中国上海)购买C57BL/6小鼠和BALB/c裸鼠(雄性,4周龄),在SPF级别动物房培养。所用动物实验的原理和实验方案均经中国科学院上海营养与健康研究所动物伦理委员会批准。
为建立荧光素酶标记的原位肝癌模型,将稳定表达荧光素酶的1×106 Hepa1-6肿瘤细胞悬浮于50μL Dulbecco’s Modified Eagle Medium (DMEM)(含20% Matrigel)中,在溴乙烷(240mg/kg, Sigma, Massachusetts,USA)麻醉下原位注射于C57BL/6小鼠肝脏。7天后,用Xenogen IVIS成像系统(Perkin Elmer,Fremont,California,USA)进行生物发光成像,评估肿瘤体积。成像前2分钟,小鼠腹腔注射10 mg/mL D-荧光素(Alameda,California,USA),剂量为150μL/只。将荷瘤小鼠随机分为对照组和不同治疗组,每5天监测一次肿瘤生长情况。
为建立皮下肝癌模型,将Hepa1-6或LPC-H12细胞(2×106在100μL(Fatal Bovine Serum)含20%Matrigel 的无FBS培养基)注入受体小鼠左侧后腿根部。荷瘤(直径约0.5cm)小鼠随机分为对照组和不同治疗组。肿瘤用游标卡尺测量,体积按公式1/2a×b2计算,其中a为长径,b为短径。
体内巨噬细胞与CD8+T细胞敲除
对于巨噬细胞敲除,根据说明书,将携带LPC-H12肿瘤的小鼠腹腔注射150μL clodronate liposomes作为第一剂量,然后每隔3天注射100μL clodronate liposomes进行更长时间的敲除。对于CD8+T细胞敲除,LPC-H12肿瘤小鼠每4天腹腔注射200μg CD8+T细胞中和抗体。对照组接受等量的生理盐水或体内MAb rat IgG2a isotype(BioXcell, West Lebanon, USA)。
骨髓来源的巨噬细胞的产生与巨噬细胞极化
取8周龄C57BL/6小鼠股骨,用加了10%FBS和20ng/ml M-CSF的 Iscove’s Modified Dulbecco’s Medium (IMDM)培养7天,去除红细胞后分离骨髓来源巨噬细胞(BMDMs,M0)。培养基每3天更换一次。为了模拟肿瘤微环境,分别于第5、6、7天将BMDM培养基从Hepa1-6改为条件培养基(CM),培养24、48、72小时(Mhepa1-6)。在无血清培养基中培养48小时后,从Hepa1-6中取CM加入新鲜BMDM培养基(比例为1:1)。对于M1极化,成熟BMDMs在含10%FBS的IMDM中与100ng/mL LPS和50ng/mL IFN-γ培养12h。对于M2极化,成熟的BMDMs在含有10%FBS的IMDM中培养24小时,其中10 ng/mL IL-4和10 ng/mL IL-13。对于CKI培养,将CKI添加到培养基中(CKI浓度:0.43、0.66、1.32 mg/mL,以CKI中总生物碱浓度为基准;培养时间:Mhepa1-6和M1为12小时,M2为24小时)。所有细胞因子均购自Peprotech(Rocky Hill, USA)。CKI中鉴定出的主要生物活性生物碱的含量见在线补充表1。
CD8+T细胞分离及增殖实验
将8周龄C57BL/6小鼠新鲜脾脏组织经70μm过滤器(BD-Falcon)获得单细胞悬液,经裂解除去红细胞。根据制造商的说明,使用EasySep小鼠CD8+T细胞分离试剂盒(Stemcell Technology,Vancouver, Canada)从单细胞悬液中分离CD8+T细胞。在CD8+T细胞增殖实验中,用carboxy fluorescein succinimidyl amino ester(CFSE,Life Technologies,San Diego,California,USA)标记CD8+T细胞,并与CKI预处理的的巨噬细胞(CD8+T:巨噬细胞=5:1,CD8+T数:2×105)在l640培养基中共培养,加入10%FBS,抗CD3(2.5μg/mL,ebiocience)和抗CD28(5μg/mL,eBioscience)72小时。之后用流式细胞仪检测CD8+T细胞增殖率,用FlowJo V.10软件进行定量分析。
肿瘤细胞活力及裂解试验
对于肿瘤细胞活性的测定,Hepa1-6细胞接种在96孔板中,在加入了10%FBS的 DMEM中以5000个细胞/孔。在培养箱中培养过夜后,将CKI(0.43 mg/mL)和索拉菲尼(0、10、15或20μM)加入培养基中。3天后,用Cell TiterGlo ATP Luminescent assay kit(Promega,Madison,USA)测定细胞数。在肿瘤细胞裂解实验中,将稳定表达荧光素酶的Hepa1-6细胞接种于CD8+T细胞上,与CKI预处理的的巨噬细胞共培养,接种在96孔板1640培养基中,加入10%FBS(CD8+T:Hepa1-6=12:1,总细胞数:5000),37℃培养24小时。为了检测肿瘤细胞的溶解水平,在96孔板中加入D-荧光素(10μL/孔)。采用Fluoroskan-FL微型平板荧光计和Thermo-Fisher科学光度计对每个孔板的荧光值进行测定。
统计分析
用GraphPad V.7对数据进行分析。用Student’s two-tailed t-test检验组内差异。绘制Kaplan-Meier生存曲线,用对数秩检验进行比较。流式细胞术数据用FlowJo V.10分析。P<0.05时组间有差异。
结论
CKI增强了低剂量索拉菲尼的治疗效果,减少了肿瘤复发并引发了有效的抗肿瘤免疫记忆反应
为了研究CKI和索拉菲尼的联合作用,我们测定了低临床剂量(10mg/kg)和临床剂量(30mg/kg)索拉菲尼抗肝癌的作用。尽管30 mg/kg索拉菲尼处理后肿瘤生长被有效抑制(在线补充图1A),但索拉菲尼导致体重减轻(在线补充图1B),天冬氨酸转氨酶(ALT)和丙氨酸转氨酶(AST)显著升高(在线补充图1C),表明索拉菲尼引发了副作用,尤其是肝毒性相关副作用。相比之下,亚临床剂量的索拉菲尼(10 mg/kg)导致抗肿瘤作用减弱,体重或肝肾功能没有变化(在线补充图1B和C)。
为了开发一种更安全、更有效的治疗策略,我们在C57BL/6小鼠的原位肝肿瘤模型和两个皮下模型中评估了CKI(每只小鼠150μL)和亚临床剂量索拉菲尼联合治疗的效果。在这里,CKI单独治疗抑制肿瘤生长;然而,联合治疗在原位和皮下肿瘤中有最显著的抑制生长效果(图1A-C)。如预期的那样,联合治疗并没有导致小鼠明显的体重减轻、肝毒性或肾毒性(在线补充图2A、B和C),表明这一策略在肝癌的治疗中可能是有效的。同时,我们还比较了CKI的治疗效果,以及与不同剂量索拉菲尼联合治疗时的LPC-H12皮下肿瘤模型(在线补充图3A)。CKI联合10 mg/kg索拉菲尼显示出与临床30 mg/kg索拉菲尼相当的抗肝癌作用(在线补充图3A)。尽管CKI联合30mg/kg剂量的索拉菲尼具有最有效的抗肝癌作用(在线补充图3A),但该治疗导致ALT和AST显著升高(在线补充图3E和F),表明CKI不能降低30mg/kg索拉菲尼导致的副作用。考虑到安全性和抗肿瘤作用,我们选择CKI和10mg/kg索拉菲尼作为体内联合治疗剂量。
为了探讨CKI和索拉菲尼联合治疗对肝癌复发的影响,建立了两种皮下肝癌手术切除和治疗模型(图1D,E)。对照组的大多数小鼠在肿瘤切除后复发,而联合治疗,无论是在手术前还是手术后开始,术后复发都显著减少了(图1D,E)。此外,我们还探讨了接受联合治疗的小鼠是否对复发肿瘤有记忆反应。对荷瘤小鼠进行手术切除肿瘤,并重新植入肿瘤细胞(图1F)。尽管在第二次肿瘤植入后没有进行治疗,但在预先接受CKI联合治疗的小鼠中,移植肿瘤的生长受到抑制(图1F),这表明联合治疗导致了小鼠对HCC的长期免疫记忆。
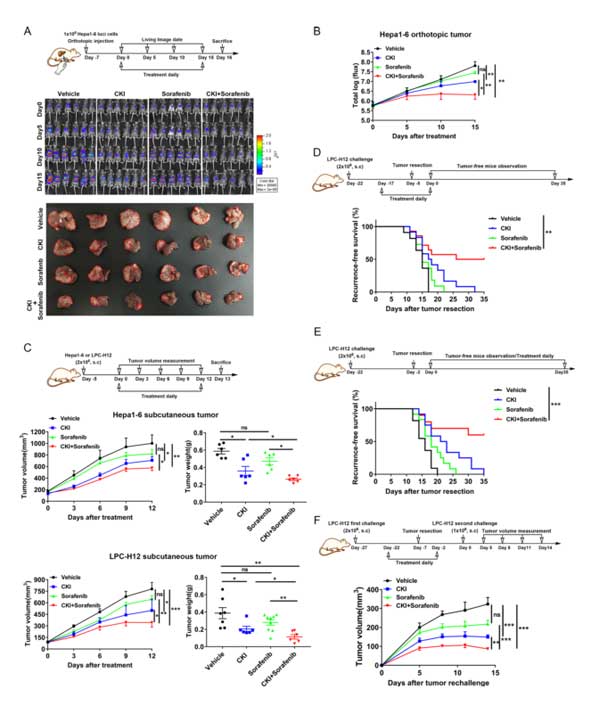
图一 复方苦参注射液(CKI)联合小剂量索拉菲尼抑制小鼠肝癌生长,减少术后复发和再种植肿瘤的生长。(A) 原位肝癌模型治疗和成像时间表(上图)。原位Hepa1-6肿瘤负荷小鼠的典型生物发光图像(中图)。小鼠每日腹腔注射CKI(150μL),灌胃索拉菲尼(10mg/kg),CKI联合索拉菲尼或对照组(n=6/组)。显示治疗后肝脏肿瘤的代表性照片;红色虚线表示肿瘤区域(下图)。(B) 治疗过程中肝脏肿瘤的相对发光变化如(A)所示。(C) 皮下肝癌模型治疗和肿瘤测量时间表(上图)。在C57BL/6小鼠的左侧分别注射2×106lpc-H12或Hepa1-6细胞(每组6-8个)。根据原位模型,每天给小鼠注射所示药物。测量肿瘤生长和肿瘤重量。(D) 手术后癌症治疗及复发观察时间表(上表)。显示无复发小鼠生存率(对照组,n=11;CKI,n=12;索拉菲尼,n=12;CKI+索拉菲尼,n=10)。(E) 手术后癌症治疗及复发观察时间表(上表)。显示无复发小鼠生存率(对照组,n=11;CKI,n=12;索拉菲尼,n=11;CKI+索拉菲尼,n=14)。(F) 手术后肿瘤再种植模型治疗和测量时间表(上图)。测定小鼠肿瘤体积(n=7)。数据以平均值±标准误差平均值。NS,p>0.05;*p<0.05;**p<0.01;***p<0.001。
CKI联合低剂量索拉菲尼降低肿瘤微环境中M2-TAMs水平,增加M1-TAMs和CD8+T的浸润
采用流式细胞仪检测CKI联合低剂量索拉菲尼对肝癌微环境的影响。在原位和皮下肿瘤中,单独或联合治疗后,总浸润免疫细胞的数量增加(在线补充图4A)。同时,单用CKI和联合治疗可显著增加M1-TAMs的比例,减少M2-TAMs(图2A,B)。Hepa1-6皮下肿瘤中的M0巨噬细胞也减少(在线补充图4B)。HCC肿瘤中CD8+ T细胞显著增加(图2A,B)。其他免疫细胞如CD4+T细胞、骨髓源性抑制细胞(MDSCs)、中性粒细胞、DC细胞和自然杀伤细胞(NK)在血液或肿瘤组织中没有受到显著影响,但联合治疗降低了荷Hepa1-6小鼠血液中的MDSCs(在线补充图4B和C)。在CKI和30 mg/kg索拉菲尼联合治疗中也观察到这些现象(在线补充图3B-D)。除治疗对免疫细胞群的影响外,功能抑制受体的表达,包括淋巴细胞活化基因3(Lag-3)、程序性细胞死亡蛋白1(PD-1)、具有免疫球蛋白和免疫球蛋白结构域的T细胞免疫受体(TIGIT)和含有免疫球蛋白和粘蛋白结构域的T细胞免疫受体(Tim-3),对CD8+T细胞也进行了测定(图2C)。与其他治疗方法相比,联合治疗有效地降低了CD8+T细胞表面功能抑制受体(图2C)。这些结果提示CKI和联合治疗通过巨噬细胞和CD8+T细胞导致肿瘤微环境的重塑。值得注意的是,在索拉菲尼治疗的小鼠中没有观察到免疫调节作用,这与索拉菲尼直接靶向癌细胞的事实是一致的。在免疫组化检测中,CKI或联合治疗组的肿瘤组织显示M2-TAMs(Arg-1)下降,M1-TAMs(INOS)和CD8+T细胞增加(图2D)。此外,裂解的caspase-3和末端脱氧核苷酸转移酶介导的dUTP缺口末端标记(TUNEL)染色增强(图2D),表明CKI联合治疗促进了体内HCC细胞的凋亡。
巨噬细胞和CD8+T细胞参与CKI和联合治疗的抗肝癌活性
为了探讨对免疫微环境的作用,我们观察了CKI和联合治疗对裸鼠肝癌的治疗作用。在任何组中均未观察到肿瘤抑制(在线补充图5A)。在体外,我们研究了CKI治疗是否对CD8+T细胞增殖有明显影响,发现CKI对CD8+T细胞增殖没有影响(在线补充图5D)。此外,CKI没有增强索拉菲尼对体外肝癌细胞的细胞毒性(在线补充图5E)。尽管裸鼠缺乏T细胞,但联合治疗后,肿瘤中的M1-TAMs仍然显著增加,M2-TAMs显著减少(在线补充图5B)。单用CKI并没有增加M1-TAMs比值,但CKI治疗后M1/M2的倍数变化仍上调,说明CKI或联合治疗仍能改善肿瘤微环境(在线补充图5B和C)。这些结果表明,CKI通过免疫调节而非细胞毒性机制提高了索拉菲尼的治疗效率。
我们进一步用CD8中和抗体阻断肝癌C57BL/6小鼠CD8+T细胞。在此,我们首先评估CD8a中和抗体的CD8+T细胞敲除效率(在线补充图6A)。正如预期的那样,流式结果表明,抗CD8抗体有效地清除了CD8+T细胞,而小鼠血液和脾脏中单核细胞或巨噬细胞的比例没有改变(在线补充图6B)。然后,我们发现CD8+T细胞的敲除消除了CKI和索拉菲尼联合治疗的抗肝癌作用(图3A),证实了CD8+T细胞在联合治疗中的作用。阻断CD8+T细胞后,尽管肿瘤内和血液中CD8+T细胞的数量较低,但通过肿瘤组织的联合治疗,M1-TAMs仍显著增加,M2-TAMs仍显著减少(图3B和在线补充图7B),而其他类型的免疫细胞在小鼠肿瘤或血液中均未发生改变(在线补充图7A和B)。经CD8+T细胞耗竭联合治疗后,M1/M2倍变化仍上调(在线补充图7C),提示CD8+T细胞介导了抗肿瘤作用,而不是联合治疗的巨噬细胞免疫调节作用。此外,免疫组化检测也表明,CD8+T细胞的缺失并不影响联合治疗对M2-TAMs和M1-TAMs的影响(图3H)。基于这些结果,我们推测CKI通过激活HCC微环境中的巨噬细胞间接增强CD8+T细胞的功能。
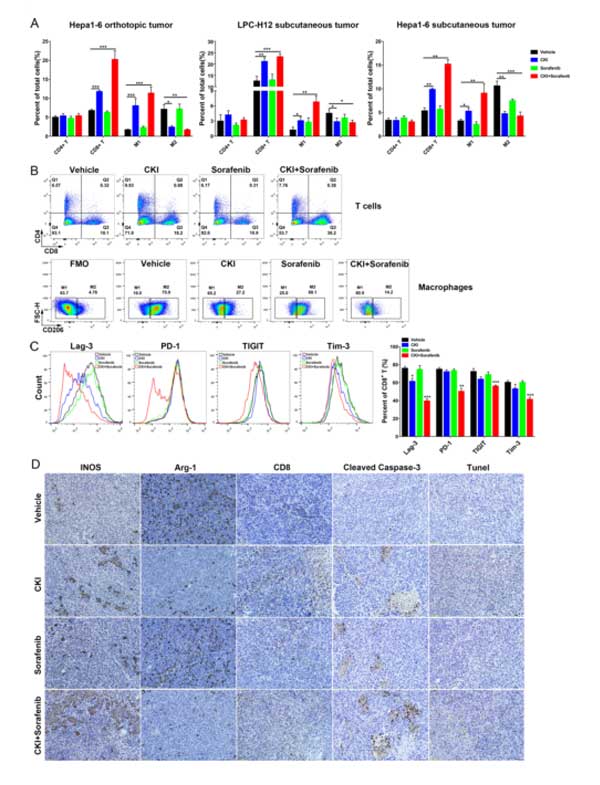
图2、复方苦参注射液(CKI)联合小剂量索拉菲尼降低M2-TAMs的分布,增加M1-TAMs和CD8+T细胞在肿瘤微环境中的比例。(A) 用流式细胞仪(FACS)测定治疗后肿瘤浸润免疫细胞在小鼠肿瘤组织中的比例。(B) 具有代表性的流式细胞术门控图像显示小鼠原位Hepa1-6肿瘤组织中CD8+T细胞、CD4+T细胞、M1-TAMs和M2-TAMs的百分比。(C) 流式细胞术检测CD8+T细胞功能衰竭标志物Lag-3、PD-1、TIGIT和Tim-3的表达。(D) LPC-H12肿瘤切片中INOS、Arg-1、CD8、caspase-3和TUNEL染色均为典型的免疫组织化学染色。数据以平均值±标准误差平均值表示。*p<0.05;**p<0.01;***p<0. 001。Arg-1,精氨酸酶1;INOS,诱导型一氧化氮合酶;Lag 3,淋巴细胞活化基因3;PD-1,程序性细胞死亡蛋白1;TAMs,肿瘤相关巨噬细胞;TIGIT,具有Ig和ITIM结构域的T细胞免疫受体;Tim-3:T细胞免疫球蛋白和粘蛋白结构域,含-3;TUNEL,末端脱氧核苷酸转移酶介导的dUTP-nck末端标记。

图3、 巨噬细胞或CD8+T细胞的敲除消除复方苦参注射液(CKI)联合小剂量索拉菲尼的抗肝癌作用。(A) 将LPC-H12皮下肿瘤C57BL/6小鼠(n=7-8)注射中和性抗CD8抗体,去除CD8+T细胞。药物治疗后,测量肿瘤生长、肿瘤重量和体重。(B) FACS检测CD8+T细胞缺失性肿瘤中CD4+T、CD8+T、M1-TAMs和M2-TAMs的比例。(C) C57BL/6小鼠(n=7)皮下接种LPC-H12肿瘤细胞,用氯膦酸脂质体清除巨噬细胞。治疗后测量肿瘤生长、肿瘤重量和体重。(D) 用流式细胞仪(FACS)测定巨噬细胞敲除后肿瘤中CD4+T、CD8+T、M1-TAMs和M2-TAMs的比例。(E) Clodronate liposome对C57BL/6巨噬细胞敲除后进行巨噬细胞补充示意图。(F) 观察不同治疗组巨噬细胞补充后肿瘤生长和肿瘤重量的变化。(G) 治疗后巨噬细胞补充肿瘤组织中CD4+T和CD8+T细胞的比例。(H) CD8+T细胞敲除后的LPC-H12皮下肿瘤中INOS、Arg-1和CD8的免疫组化染色。(I) 巨噬细胞敲除后LPC-H12皮下肿瘤中INOS、Arg-1和CD8的免疫组化染色。数据以平均值±标准误差平均值表示。NS,p>0.05;*p<0.05;**p<0.01;***p<0. 001。Arg-1,精氨酸酶1;INOS,诱导型一氧化氮合酶。
接下来,我们用clodronate liposomes对荷皮下LPC-H12肿瘤的C57BL/6小鼠进行了巨噬细胞在C57BL/6小鼠中的作用研究。在小鼠血液和脾脏中,clodronate liposomes对单核细胞或巨噬细胞有有效的敲除,而对CD8+T细胞没有影响(在线补充图6A和B)。巨噬细胞敲除能够抑制肿瘤生长,这一发现支持肿瘤浸润巨噬细胞在肿瘤发展中的促进作用(图3C)。然而,在巨噬细胞敲除后的小鼠中,CKI和索拉菲尼联合治疗不再抑制肿瘤的生长(图3C)。同样,敲除后,对照组和治疗组的肿瘤微环境或小鼠血液中的M1-TAMs、M2-TAMs或单核细胞比例仍然较低,而CKI和索拉菲尼联合治疗组的CD8+T细胞比例也没有升高(图3D和在线补充图7B)。在巨噬细胞敲除后的肿瘤中,联合治疗不能诱导肿瘤内CD8+T细胞增加(图3D,I),证实了巨噬细胞对CD8+T细胞浸润的必要性,而其他类型的免疫细胞在小鼠肿瘤或血液中没有改变(在线补充图7A和B)。在此,我们还发现联合治疗增加的M1/M2比率被巨噬细胞敲除所消除(在线补充图7C),表明巨噬细胞敲除也消除了联合治疗对M2到M1转变的影响。为了进一步证实联合治疗后巨噬细胞在增强CD8+T细胞抗肿瘤功能的作用,我们在巨噬细胞敲除后的小鼠中建立巨噬细胞回输的肿瘤模型(图3E)。虽然巨噬细胞敲除后小鼠的肿瘤生长较慢,但在巨噬细胞补充后,巨噬细胞敲除引起的肿瘤生长下降得以恢复(在线补充图8),这表明补充组与对照组相比观察到的肿瘤生长是由巨噬细胞介导的。重要的是,联合治疗进一步抑制了敲除后补充的巨噬细胞所介导的肿瘤生长(图3F)。相应地,联合治疗增加了巨噬细胞敲除时CD8+T细胞的浸润(图3G)。因此,CKI通过降低M2-TAM的分布,提高M1-TAMs的水平,进而激活CD8+T细胞增强抗肿瘤免疫,增强了索拉菲尼的抗肝癌作用。
CKI诱导TAMs向M1极化并增强促炎作用
在HCC微环境中,巨噬细胞可以被激活到不同的状态,包括肿瘤杀伤M1激活和肿瘤促进M2激活11。由于CKI调节了M1-TAMs和M2-TAMs在HCC微环境中的比例,我们确定了CKI治疗对巨噬细胞极化的影响(图4A)。首先,我们通过与Hepa1-6 细胞培养上清(CM)共培养24、24和72小时来评估Hepa1-6 CM是否能使BMDMs极化到免疫抑制状态,以模拟HCC微环境(在线补充图9A)。结果表明,随着Hepa1-6 CM共培养时间的延长,BMDMs极化到更抗炎的状态,对CFSE标记的CD8+T细胞的增殖具有更强的免疫抑制作用(在线补充图9A-D)。然后将Hepa1-6 CM培养的BMDMs(Mhepa1-6)与不同浓度的CKI体外培养。流式细胞仪分析显示,CKI使峰值向M1极化移动,导致M1巨噬细胞显著增加,M2巨噬细胞呈剂量依赖性减少(图4B)。此外,在CKI治疗后,M1标记物和促炎因子的mRNA水平增加,而M2标记物(抗炎因子)的表达水平降低(图4C和在线补充图10)。M1和M2相关炎症细胞因子或蛋白质的浓度也有相同的趋势(图4D)。这些结果表明CKI诱导巨噬细胞向M1分化。
接下来,我们研究了CKI对极化巨噬细胞的影响。将BMDMs诱导成M1或M2状态,然后加入不同剂量的CKI。采用qPCR法和ELISA法检测典型炎症因子的表达。在M1和M2巨噬细胞中,CKI以剂量依赖的方式增强促炎基因,下调抗炎因子(图4E-H),表明CKI促进巨噬细胞向促炎极化转变。为了在体内验证这一发现,我们分离了小鼠肝癌肿瘤负荷中的肿瘤浸润巨噬细胞,并测量了炎症因子的水平。在接受CKI和联合治疗的小鼠中,肿瘤浸润巨噬细胞中的促炎因子显著升高,抗炎因子被抑制(图4I)。
CKI刺激后的巨噬细胞促进CD8+T细胞的增殖和杀伤活性,减轻其功能耗竭
为了探讨CKI预处理的巨噬细胞对CD8+T细胞的影响,原代CD8+T细胞与CKI预处理的Mhepa1-6、M1或M2巨噬细胞共培养(图5A)。我们发现,尽管CKI已经被移除,CKI处理后的Mhepa1-6、M1和M2巨噬细胞依然促进CD8+T细胞的增殖(图5B和在线补充图9C和E)。重要的是,CKI的培养以剂量依赖的方式释放Mhepa1-6和M2巨噬细胞对CD8+T细胞增殖的免疫抑制作用(图5B和在线补充图9C和E)。接下来,我们测量了CD8+T细胞的细胞毒性。与CKI诱导的Mhepa1-6、M1或M2巨噬细胞共培养后,CD8+T细胞中典型细胞毒素细胞因子的基因表达和蛋白分泌均上调(图5C、D)。为了研究CKI诱导的巨噬细胞如何作用于CD8+T细胞(通过接触依赖效应或巨噬细胞产生可溶性分子介导),收集CKI预处理的Mhepa1-6细胞培养上清并与CD8+T细胞共培养(在线补充图11)。CKI-Mhepa1-6上清液诱导CD8+T细胞中细胞毒素细胞因子的增殖水平与CKI-Mhepa1-6相似(在线补充图11),表明巨噬细胞产生的可溶性分子负责CKI预处理的巨噬细胞对CD8+T细胞的功能。我们用CKI预处理(处理后去除CKI)后的巨噬细胞与CD8+T细胞共培养,分离后的CD8+T细胞与Hepa1-6细胞共培养检测CD8+T细胞的肿瘤细胞杀伤力。CKI治疗组Hepa1-6细胞的溶解率显著增加(图5E),表明CKI预处理的巨噬细胞显著改善了CD8+T细胞的细胞毒性功能。此外,在先前与CKI预处理的巨噬细胞共培养的CD8+T细胞中,典型的功能耗竭特异性基因水平降低(图5C)。CD8+T细胞的细胞毒性升高和功能耗竭缓解在体内得到证实(图2C和5F)。值得注意的是,尽管CKI和联合治疗HCC肿瘤的CD8+T细胞中的细胞毒性标记物增加,但联合治疗组的所有CD8+T细胞衰竭标记物数量均降低,而CKI单独治疗组的Lag-3和Tim-3数量均降低(图2C和5F),表明索拉菲尼可能有助于缓解体内CD8+T细胞功能耗竭。这些结果可以部分解释为什么CKI联合索拉菲尼治疗可以降低肿瘤复发率,并对HCC产生有效的抗肿瘤记忆反应。
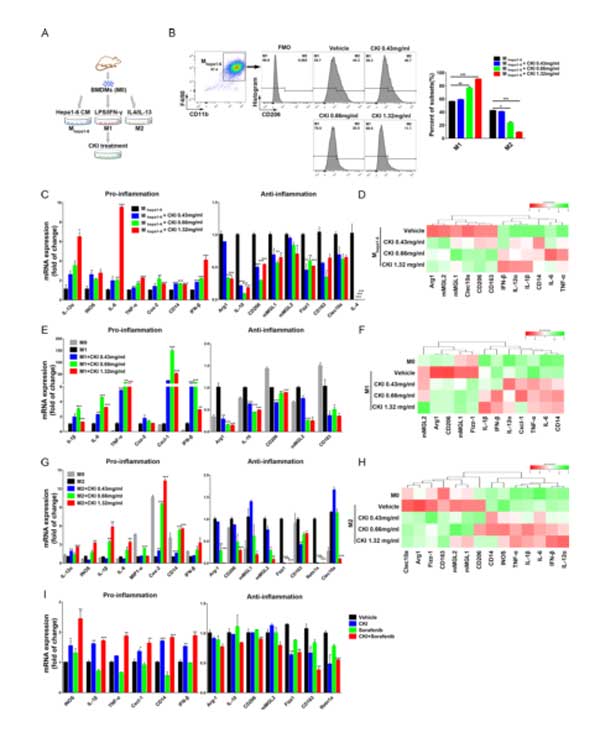
图4、复方苦参注射液(CKI)能促进荷瘤巨噬细胞极化至M1状态,增强促炎功能,降低抗炎功能。(A) 骨髓来源巨噬细胞(BMDMs)治疗后不同极化状态示意图条件培养基(CM)24小时(Mhepa1-6)、100 ng/mL LPS和50 ng/mL IFN-γ治疗12小时(M1)或10 ng/mL IL-4和10 ng/mL IL-13治疗24小时(M2),然后进行CKI治疗。(B) 将Mhepa1-6与不同浓度的CKI培养12h。流式细胞仪测定M1和M2巨噬细胞的比例。(C) 采用定量实时聚合酶链反应(qRT-PCR)检测不同剂量CKI治疗12h后Mhepa1-6中M1(促炎症)和M2(抗炎)巨噬细胞标志物的表达。(D) 用ELISA法测定CKI诱导Mhepa1-6细胞培养上清或细胞裂解液中M1、M2相关细胞因子或蛋白的含量。(E和F)BMDMs(M0)极化至M1状态,用不同剂量的CKI治疗12小时。用qRT-PCR(E)或ELISA(F)检测促炎和抗炎基因的表达。(G和H)BMDMs(M0)极化至M2状态,用不同剂量的CKI治疗24小时。用qRT-PCR(G)或ELISA(H)检测促炎和抗炎基因的表达。(I) 治疗后,从LPC-H12小鼠皮下肿瘤中分离肿瘤相关巨噬细胞。qRT-PCR检测促炎和抗炎基因mRNA水平。数据以平均值±标准误差平均值表示。*p<0.05;**p<0.01;***p<0.001。

图5、复方苦参注射液(CKI)诱导的巨噬细胞能提高CD8+T细胞的增殖和杀伤能力,缓解其功能耗竭。(A) CKI诱导巨噬细胞与CD8+T细胞共培养系统(巨噬细胞:CD8+T比值=5:1,CD8+T数=2×105)与抗CD3(2.5μg/mL)和抗CD28(5μg/mL)刺激72小时示意图。(B) 用羧荧光素琥珀酰氨基酯(CFSE)标记CD8+T细胞,与CKI诱导的巨噬细胞(Mhepa1-6、M1和M2)共培养72h。用CD3/CD28抗体刺激CD8+T细胞,与载体或CKI诱导的巨噬细胞共培养(左面板),绘制CD8+T细胞CFSE信号强度的典型图。计算各组(右面板)CD8+T细胞增殖的数量。(C) 定量实时PCR(qRT-PCR)检测CKI诱导的巨噬细胞(Mhepa1-6、M1和M2)共培养CD8+T细胞中细胞毒和耗尽标记物的表达。(D) ELISA法测定共培养上清中细胞毒细胞因子的含量。(E) 用荧光素酶标记的Hepa1-6细胞共培养CD8+T细胞24小时(CD8+T:Hepa1-6比值=12:1,细胞总数=5000)。荧光法检测Hepa1-6肿瘤细胞的溶解程度。(F) 用上述方法从LPC-H12小鼠皮下肿瘤中分离CD8+T细胞。用qRT-PCR法检测CD8+T细胞中细胞毒基因和耗尽基因的mRNA水平。数据以平均值±标准误差平均值表示。*p<0.05;**p<0.01;***p<0.001。
CKI通过巨噬细胞表面TNFR1受体及其下游NF-κbp65和MAPK p38信号通路激活巨噬细胞促炎功能
巨噬细胞在GM-CSF、IFN-β、IFN-γ、LPS或TNFα的刺激下,通过相应的受体CSF2Rα、IFN-α/βR、IFN-γR、TLR4或TNFR1极化成M1促炎状态26 27。为了阐明CKI对巨噬细胞的作用,我们检测了这些受体在肝癌组织TAMs中的表达。在接受CKI治疗的小鼠中,TAMs显示IFN-γR、TLR4和TNFR1的表达上调(图6A)。这一发现在CKI启动的Mhepa1-6、M1和M2巨噬细胞中得到证实(图6B和在线补充图12A)。为了探讨CKI对巨噬细胞的引导作用,我们用R-7050、Sparstolonin B和抗IFN-γR抗体阻断Mhepa1-6细胞中的TNFR1、TLR4或IFN-γR。TLR4或IFN-γR阻断不影响巨噬细胞中CKI介导的促炎症功能和TNFR1上调(在线补充图12B和C),而CKI诱导的促炎细胞因子和TNFR1的升高是通过TNFR1阻断(图6C和在线补充图12C)预防的,这表明CKI通过靶向TNFR1诱导巨噬细胞。
在此基础上,我们确定了CKI对TNFR1激活及其下游信号传导的影响。流式细胞术分析显示,CKI促进增加了Mhepa1-6细胞表面TNFR1的表达(图6D)。除此之外,TNFR1与TRADD、RIP1和TRAF2的关联性增强(图6E),表明CKI促进了TNFR1活化所必需的蛋白质复合物的形成。TRAF2的蛋白质水平,最终连接TNFR1复合物以触发下游信号,也被上调(图6E)。这些结果表明CKI增强了TNFR1信号的激活。同样,CKI促进后,Mhepa1-6中p-TAK1、p-IKKα/β、p-IκBα、p-P65和p-P38的诱导量更高(图6F),而CKI治疗对JNK和ERK的影响较小(在线补充图12D)。当细胞表面TNFR1被抗TNFR1抗体中和或TNFR1/TRADD/RIP1复合物的形成被R-7050抑制时,CKI诱导的TRAF2、p-TAK1、p-P65和p-P38的增强表达也被阻断(图6G)。
CKI通过TNFR1对亚临床剂量索拉菲尼治疗肝癌的增敏作用
在CKI和索拉菲尼联合治疗期间,将LPC-H12皮下肿瘤小鼠腹腔注射抗TNFR1抗体或R-7050。抗TNFR1抗体或R-7050消除了CKI联合索拉菲尼的抗肝癌作用(图6H和在线补充图12E),表明TNFR1介导了CKI的治疗作用。综上所述,CKI作用于巨噬细胞,促进TNFR1与TRADD/RIP1/TRAF2的结合,并触发下游NF-κB和MAPK p38通路,发挥促炎作用,促进TAM向M1-TAM-极化,并逆转M2-TAMs的极化。CKI处理后的巨噬细胞进一步促进CD8+T细胞的增殖和杀伤活性,诱导肝癌细胞凋亡,同时降低CD8+T细胞功能性耗竭,维持抗肝癌免疫记忆活性,最终抑制肝癌的生长和复发。由于两种药物相互促进的抗肿瘤机制,将CKI与亚临床剂量的索拉菲尼联合治疗可以在提高安全性的同时大大增强抗肝癌的疗效(图7)。
讨论
CKI作为一种中药复方,已被NMPA批准用于治疗癌症引起的疼痛17。CKI在乳腺癌19、结肠癌20、非小细胞肺癌21、急性白血病22、肝癌23-25中的治疗也进行了研究。索拉菲尼作为一线药物,长期以来被广泛应用于晚期肝癌的治疗。然而,由于严重的副作用、获得性耐药、肿瘤异质性和免疫抑制微环境,只有30%的肝癌患者能从索拉菲尼中获益。在这项研究中,我们结合CKI和亚临床剂量的索拉菲尼治疗肝癌。这项新战略代表着重大进展。首先,降低索拉菲尼的剂量,可以大大减少索拉菲尼的不良反应,从而克服毒性问题。其次,CKI调节肿瘤微环境,索拉菲尼直接靶向肿瘤细胞。这些不同的作用方式促进了抗癌活性,并取得了显著的抑瘤效果。第三,CKI通过免疫记忆保护CD8+T细胞对抗HCC,从而防止复发,比单独使用索拉菲尼获得更好的预后。巨噬细胞或CD8+T细胞的敲除终止了联合治疗的抗癌作用,证实了这些免疫细胞在治疗中的重要性。
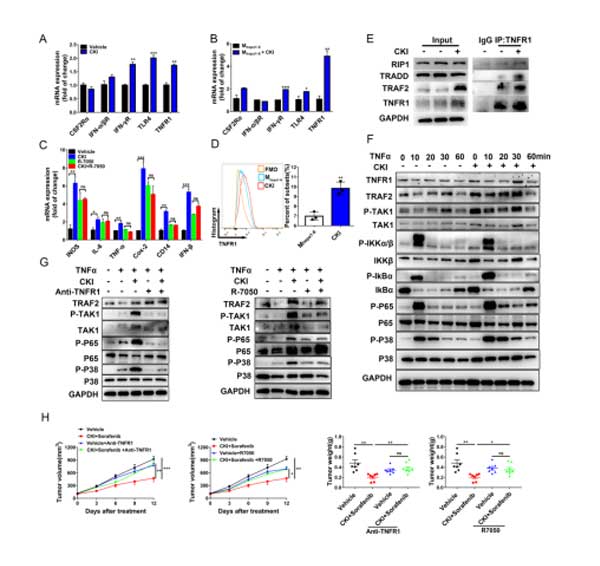
图6、复方苦参注射液(CKI)促进TNFR1复合物相互作用和NF-κbp65、MAPK p38信号传导,发挥促炎作用。(A) 以LPC-H12小鼠皮下肿瘤为研究对象,采用定量实时PCR(qRT-PCR)方法检测促炎性极化相关受体基因mRNA水平。(B) 用0.66mg/mL CKI培养12h,检测Mhepa1-6中促炎性极化相关受体的表达。(C) 测定了TNFR1阻断剂(R7050,10μM)作用于0.66mg/mL CKI 12h后Mhepa1-6中促炎和抗炎基因的表达。(D) 用流式细胞仪(FACS)测定0.66mg/mL CKI-Mhepa1-6中TNFR1的细胞表面水平。(E) 用0.66mg/mL CKI处理Mhepa1-6细胞12h,用免疫共沉淀法检测TNFR1与TRADD、RIP1和TRAF2的结合情况。(F) Mhepa1-6细胞经0.66mg/mL CKI作用12h,10ng/mL TNF-α作用不同时间,western blotting检测蛋白表达。(G) 用25μG/mL抗TNFR1抗体(左板)或10μM R7050(右板)处理Mhepa1-6 4h,再用0.66mg/mL CKI处理12h,然后用10ng/mL TNF-α刺激30min,western blotting法测定TRAF2、磷酸化和总TAK1、P65和P38的表达量。(H) 皮下LPC-H12荷瘤小鼠每日用CKI和索拉菲尼(CKI,150μL;索拉菲尼,10 mg/kg)、抗TNFR1抗体(100μg/只小鼠,每4天)、R-7050(10 mg/mL,每3天)或载体(n=7/组)治疗。测量肿瘤生长和肿瘤重量。数据以平均值±标准误差平均值表示。NS,p>0.05;*p<0.05;**p<0.01;***p<0. 001。TNF-α、肿瘤坏死因子α、TNFR-1、肿瘤坏死因子受体超家族1。

图7、复方苦参注射液(CKI)重建肝细胞癌(HCC)微环境及其与小剂量索拉菲尼联合应用的疗效。INOS,诱导型一氧化氮合成酶;IFN-β,干扰素- β;IL,白细胞介素;Lag-3,淋巴细胞活化基因3;Tim-3,T细胞免疫球蛋白和粘液蛋白结构域,含-3;TIGIT,T细胞免疫受体,含Ig和ITIM结构域;TNF-α,肿瘤坏死因子α。
肝癌具有侵袭性,有效的治疗方法仍然有限4。免疫疗法已被作为癌症治疗的一种选择研究30。有希望激活治疗性抗肿瘤免疫的方法被设计成扩大细胞毒性CD8+T细胞向肿瘤微环境的浸润,并抑制免疫抑制分子如CTLA-4、PD-1、Tim-3、Lag-3和TIGIT,也称为免疫检查点阻断31-33。其中,抗CTLA-4抗体,ipilimumab,以及两种抗PD-1抗体,Pembrolizumab和Nivolumab,已经被FDA批准用于治疗癌症32。然而,对于某些类型的肿瘤,由于免疫抑制微环境的复杂性,单以CD8+T细胞为靶点在临床上不足以用于肿瘤治疗34。
肝癌微环境形成了一种抗炎基质,并引起了MDSCs和TAMs等免疫抑制性免疫细胞,它们阻断抗原递呈过程,直接抑制CD8+T细胞的增殖和细胞毒性35。值得注意的是,TAMs是肝癌微环境中的主要免疫抑制细胞36。基于TAM的抗肝癌治疗包括抑制巨噬细胞募集、抑制TAM存活、增强TAMs的M1抑瘤活性和阻断M2促瘤活性13 36 37。结果表明,CKI和sorafenib联合治疗降低了M2-TAMs的比例和极化,增强了M1-TAMs在肿瘤微环境中的分布和极化。在Hepa1-6cm培养的巨噬细胞中,CKI剂量依赖性地增加M1-TAM特异性细胞因子(IL-12α、INOS、IL-1b、IL-6、TNF-α、Cox-2、CD14和IFN-β)的产生,并消除与M2-TAM相关的因子(Arg-1、IL-10、CD206 mMGL1、mMGL2、Fizz-1、CD163、Retn1a和Clec10a)。此外,CKI诱导的巨噬细胞也减轻了CD8+T细胞的免疫抑制作用。在巨噬细胞和CD8+T细胞共培养体系中,CKI预处理后的巨噬细胞能促进CD8+T细胞的增殖,揭示了CD8+T细胞的活化。CKI诱导的巨噬细胞增强了肿瘤细胞的细胞毒性因子分泌(TNF-α、穿孔素、颗粒酶B和IFN-γ)和CD8+T细胞对肿瘤细胞的杀伤作用,意味着抗肿瘤细胞毒活性已被重新唤醒。
此外,在慢性抗原暴露和炎性条件下,T细胞无法执行效应器功能,这种状态称为T细胞功能性耗竭38。肿瘤微环境中的大多数T细胞已耗竭31,T细胞功能性耗竭可由多种细胞群诱导,包括调节性T细胞、M2-TAMs、MDSCs和肿瘤细胞32 39-41。在我们的研究中,在CKI和索拉菲尼联合体内治疗或体外与CKI诱导的巨噬细胞共培养后,CD8+T细胞的耗竭状态得到缓解,表现为抑制性受体(Lag-3、PD-1、TIGIT和Tim-3)水平降低。激活的免疫系统可引起肿瘤抗原特异性记忆T细胞的持久性,从而对肿瘤复发作出有效的记忆反应32。同理,在我们的术后复发模型和肿瘤再种植模型中,CKI和索拉菲尼联合治疗可抑制肿瘤的形成和生长。由于CKI单独或联合治疗不能完全根除肿瘤生长,肿瘤的微环境复杂,即使联合治疗也有可能发生更多的HCC复发。
NF-κB途径是肝癌产生的一个中心调控因子42。在肿瘤发生的早期,M1巨噬细胞NF-κB途径的激活对肿瘤相关炎症的发生至关重要43。然而,在肿瘤发生的晚期,TAMs在肿瘤微环境中极化成M2,表现为低NF-κB途径激活,但免疫抑制能力增强44。因此,M2-TAMs转换成M1-TAMs以促进NF-κB途径激活是肿瘤微环境重塑的一个有前途的策略44。我们发现CKI在体内和体外可以通过TNFR1/NF-κB和MAPK轴在巨噬细胞中将M2转换成M1状态并发挥促炎作用,为CKI通过TAMs逆转TAM介导的免疫抑制提供了证据。
综上所述,CKI降低了M2-TAMs的浸润和极化,提高和促进了肿瘤微环境中M1-TAMs的比例和极化,从而减轻了对CD8+T细胞的免疫抑制作用,最终增强了CD8+T细胞抗肿瘤细胞的毒性功能和抗肿瘤记忆作用。CKI联合低临床剂量索拉菲尼能有效抑制肝癌的生长和复发,且无明显毒性。我们的发现提供了临床前证据,表明中药联合化疗可能在人类肝癌治疗中具有重要意义,并且CKI是一个有前途的候选药物。
基金资助
这项工作得到了国家重点研发计划(2018YFC200700)、国家自然科学基金委员会(81630086和81973078)、中国科学院重点研究项目(ZDRW-ZS-2017-1)、上海市卫生委员会(2017YQ0059),上海市人力资源和社会保障局(2018060)、上海市教委重大科技创新计划(2019-01-07-00-01-E00059)、上海交通大学(YG2017MS85)的资助。


移动平台
微信平台
